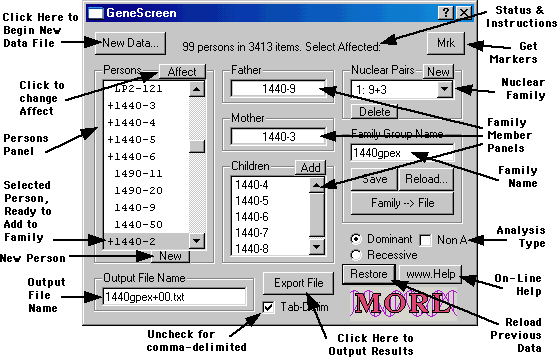
The basic elements of the GeneScreen software are evident in this annotated screen shot:
Marker data lines have three fields. The first column (before the first tab) must be a chromosome number (less than 30) or the letter "X", or else empty, in which case the previous good chromosome number is used.
| Chr | Marker | cM |
| 1 | D1S468 | 4.22 |
| 1 | D1S214 | 14.04 |
| 1 | ... |
The marker name may be in any column after that, but each marker name must start with "D" and contain an "S" followed by a unique (for that chromosome) numeric marker reference number greater than zero. Extra blank spaces are ignored. Acceptable column headers are "Marker" or "STRP", but these are advisory only. If a valid marker name is found in a different column, it still works.
The distance number may also be in any column after the first, but each distance item should be a pure number, typically with a decimal point. Distance numbers must not be negative, nor greater than 9999.99; a maximum of only two digits after the decimal point are considered, with any extra digits rounded off. The advisory column header is "cM".
Only the first item recognized respectively as a marker name or distance is accepted. Once both the marker name and the distance have been found on a line, the rest of the line is ignored. If there is only a distance on the line, it is assumed to be the distance for the end of the chromosome.
GeneScreen remembers the name and location of the last successful marker
file, and starts up reading that file again. However, you may direct it
to read a different marker file at any time, after which it will remember
the new file.
Marker -- This column should have marker names matching those in the marker file.
Sample Name -- This column should have reference names of the persons from this data was collected. GeneScreen assumes the name is in the form Family-person (separated by a single hyphen), but almost any string of characters is acceptable (commas, spaces, and plus are not allowed). The family name can be used to separate out family groups (see below). The person name is used in the output. It is recommended that person names be unique, but GeneScreen only requires uniqueness in the family-person combination. Each unique person is added to the Persons field in the main window, from which it can be added to families and/or marked as affected.
Allele 1 / Allele 2 -- These two columns contain the allele data for each person. Alleles are required to be decimal numbers greater than 0 and less than 30.
GeneScreen reads the entire data file into memory, then picks out the significant data. If it encounters a marker name that is not in the marker file, it will complain and offer to ignore that data. It is expected that there will be data lines with no allele data; these are effectively ignored. When there are multiple non-blank data lines for the same person and marker, only the first is retained.
Additional persons may be added to the Persons list by clicking the New button. GeneScreen chooses a number not already used and adds it to the end of the list. Of course there will be no data for that person, but GeneScreen can sometimes infer the data from the family structure and other family members for which there is data. If there is already a family group name, that name will be prefixed to the new person names.
After the file has been read into memory, the persons are extracted and listed in the Persons field of the window. If there is already family data active, it is reconciled to the data. It is recommended that if you want to start over with new raw data and new families, you should quit and restart GeneScreen to prevent confusion.
After the data file has been read in and displayed, you may designate
persons in the file as Affected by selecting them one at a time and clicking
the Affect button. This puts a "+" symbol to the
left of the person's name. Clicking the Affect button a second time
removes the designation.
Once constructed, you can Save the current family structure to a "familyname.fam" file. Unless you designate otherwise, the family name part of the first family member you select will be offered as a suggested Family Group Name, but you can change it to anything you like. You may Reload a saved family, make additional changes, and then save the new version for later use.
The saved family file also records whether you have the Dominant or Recessive butten checked, and which persons are affected; these are restored when the file is reloaded. If the family being reloaded contains persons not listed in the Persons field, you will be asked if you want them added.
Clicking the Restore button will quickly reload the last data
file read in and the last saved (or reloaded) family file, to make it easier
to take up where you left off on the previous day.
When you open a raw data file, GeneScreen generates a suggested output file name from the data file name. Similarly, when you load in a family data file, its name is offered as an output file name; you can also do this manually by clicking on the Family->File button. If you subsequently make changes to the family group name, the output file name will try to track the changes.
The actual output files are written when you click the Export File
button, after doing the processing. The processing consists of first filling
in the missing alleles for any persons that GeneScreen is able to infer,
then adjusting the allele order to reduce crossovers, then looking for
runs of dominant or recessive alleles. The output is in a Tab-Delimited
file (or comma-delimited, if the checkbox is unchecked).
0. Arranging the Persons (once only, for all chromosomes in file)
GeneScreen tries to place all persons in parentless families, and all persons who are parents but not children in any families, into the first row. Persons otherwise eligible for this first line, but also a parent of a family with another person who is not eligible for the first line, will be delayed to be placed next to that other parent.
After any or both parents of a nuclear family have been placed on a row, the children of that family are placed on the next row, if they will fit.
Inbred families (brother+sister, or cousins) will have the respective brother/sister or grandparent pair arranged in their respective family so that the father of the inbred family occurs on the left. Otherwise families are more or less placed in the order they occur in the family file, which is the order they were created.
Persons are not yet aligned vertically with the parents over the children, but that is planned for a future revision.
1. Headers
At the top of each output file is placed a couple of header lines consisting of the marker names and their relative distances (positional differences), followed by one line for each person in the family structure and the unprocessed allele data for that person. Affected persons are indicated, and gender (1 for male, 2 for female) is shown for persons who have been designated either father or mother. There is no other way to enter gender of persons, but non-parent persons can be added to fake one-parent, no-children families as father or mother to accomplish that purpose, if desired.
Markers for which there is no data at all are then removed from further processing.
2. Kids Allele Order
For all children with data in one or both parents, the alleles are ordered to reflect where they came from, or marked "X" if it cannot be determined.
3. Filling Gaps
For all missing alleles in parents, if the missing data can be reasonably inferred from available children's alleles, that data is added in quotes. These inferential choices are not always the best possible, but they sometimes represent a good start.
For each person who is designated a parent of some family, the data is searched for all families that person is parent of, and for which there is also a second parent. Multiple couplings tend to disable the process, or else give unreliable results. All the children of a single pair of parents are collected by marker, and organized into one of the following categories (separate letters 'x', 'y', 'z' etc represent different alleles for that marker):
3a. x,y or x,x -- If there is only one distinct pair, and neither parent has any alleles at all, then each is assigned one of the pair. If this person already has one of the alleles, then nothing further is done (the other parent will be separately considered). If the other parent already has one of the alleles and this parent has neither, then its blank is filled with that second allele. If all children are homozygous on this marker, then both parents are given that allele unless they already have it.
3b. x,x and y,y and possibly x,y -- If there are two different homozygous pairs, then both parents have one each. The extra mixed pair is not determinative and has no effect whether present or not.
3c. x,x and y,z and possibly x,y and/or x,z -- Each parent is known to have an x, and one each y or z. If either parent already has y or z, the decision is forced; otherwise they are assigned arbitrarily and later decided on the basis of crossover counts.
3d. x,y and x,z or x,x and x,y -- The latter being a degenerate form of the first (x=z). One parent is assumed to carry x, and the other parent both y and z. The prior presence of any of these forces the decision; otherwise they are assigned arbitrarily and later decided on the basis of crossover counts.
3e. x,w and x,z and y,w and y,z (or with x=w, x,x and x,y and x,z and y,z) -- One parent has x and y, the other w and z.
3f. x,y and y,z and x,z-- This could be the result of one homozygous parent in any single allele of the three, and the other two in the other parent. The prior presence of any of these forces all or part of the decision; otherwise they are assigned arbitrarily and later decided on the basis of crossover counts.
4. Crossover Minimization
Each heterozygous marker in a parent not a child in some other family with parent data forcing the allele order, is evaluated with both allele orders against all the children in that family, for a minimum total crossover count. In case of equal crossover counts, the minimum sum of inverse distance squares is preferred (this effectively minimizes incidence of double crossovers, which have a fairly large inverse square). Where inferred alleles have been arbitrarily assigned, these are tried in both alternations as well as each order. Where children have been marked as "X" because both parents have the same heterozygous pair, the children alleles are also measured in both orders. This makes for a lot of separate crossover count analyses, which might take several seconds or longer, depending on the number of different pairs to be tried in all possibilities.
5. Dominant Runs
If the Dominant checkbox is checked, GeneScreen looks for runs of the same alleles in all affected persons. If NonAffect is also checked, the non-affected (see Affect above) persons are also considered in looking for runs, and only runs of matching alleles appearing in affected persons and not in non-affected persons are identified. Otherwise the non-affected persons are not considered. A run is any sequence of two or more in all affected persons. Persons on the first two rows of putput are given more weight.
6. Recessive Runs
If the Recessive checkbox is checked, GeneScreen looks for runs of homozygous alleles in all persons, without regard to whether they are affected or not.
7. Annotations
Each person is output in the family tree with their alleles aligned
in a vertical column by marker order and the father's allele to the left.
When the parentage cannot be readily determined, an "X" is added in the
next column. Crossovers, where a child's allele switches from one grandparent
to the other, for each sequence are marked by "c" just outside the pair,
to the left or right. Runs are then indicated by 1 (for paternal alleles)
or 2 (for maternal alleles) or 3 (for both) in the next column to the left.
Rev. 2004 November 20